 手机访问:wap.265xx.com
手机访问:wap.265xx.com纳米抗体建模算法介绍
前 言
重链抗体(HCAb)可变区VHH长度通常小于130个氨基酸残基,因它们较小的尺寸使它们比传统抗体更容易生产(而且成本较低),且VHH可以独立于HCAb使用,具有热稳定性强,性质稳健的特点,容易根据其效用目标进行定制。因此,VHH得到了广泛的研究和应用,例如药物研发、CAR-T、癌症治疗、体内成像、病原体的检测或作为生物传感器等。
尽管已经开发出有前途的治疗策略并有产品已经上市(卡普赛珠单抗是FDA批准的第一个针对成年有获得性血栓形成血小板减少性紫癜的治疗性VHH),但抗体在制造和纯化方面仍然具有相当的挑战性。其中一个难点是如何优化VHH分子,以提高其稳定性、特异性、功能活性、表达量及进行人源化改造。因此,一个相关策略是预先使用计算的方法提高优化过程的效率和成功率,而计算的关键步骤是阐明分子的3D模型。同时,建模的理论和方法也是深入地了解VHH分子特性的非常有价值的工具,为新抗体的设计提供了可能性。最近,Poonam Vishwakarma等发表了一篇专门针对VHH建模算法的综述--VHH Structural Modelling Approaches: A Critical Review [1],介绍了给定VHH序列建模的21种方法,提供了每种方法的简要解释和使用的相关例子,旨在展示模型软件程序如何对VHH的结构进行预测。在此,让我们分享一下其中的几种建模算法。
首先,作者总结了一些特定的抗体建模工具。经典的蛋白质建模工具(见图1a)可以用于抗体和VHHs。同时,由于抗体在生物技术和生物医学领域的各种影响及其多样性,因此需要专门的抗体建模工具(见图1b)。
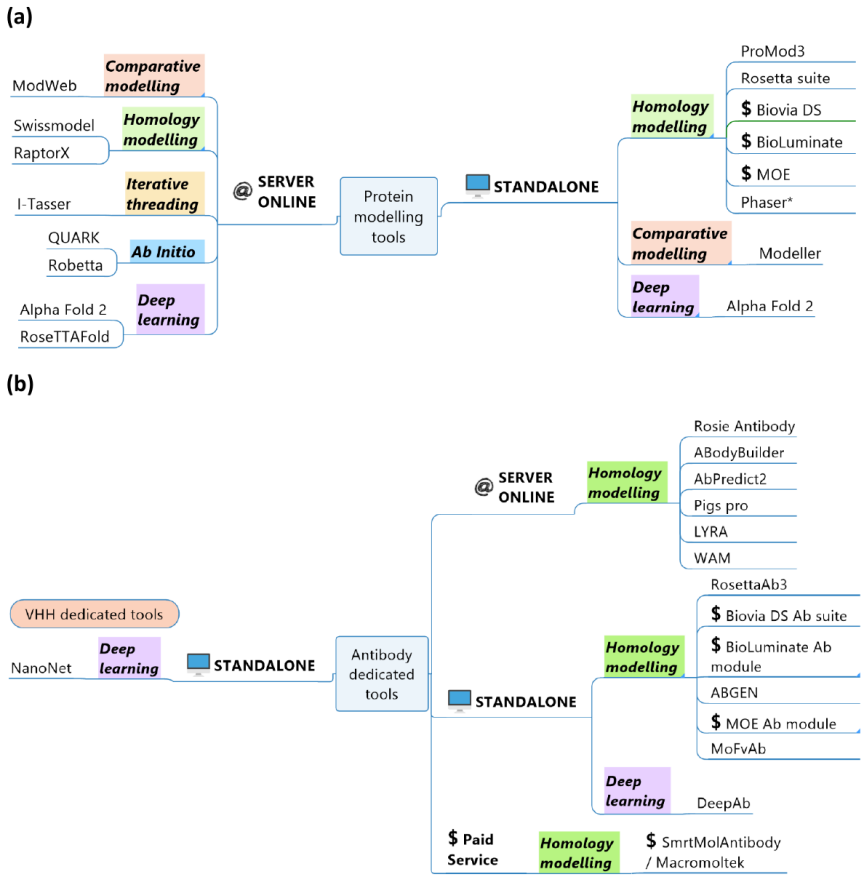
图1 蛋白质结构预测工具总结(a)一般蛋白建模工具和(b)抗体和VHH建模工具。所有的工具都根据其可用性(独立和/或在线服务器、学术或商业)和所使用的方法(比较/同源建模、线程、从头开始和深度学习)进行分类。
Modeller and Modweb
Modeller[2]可能是比较/同源性建模中最重要的软件。它开发于1993年,目前仍然维护(https://salilab.org/modeller/),对学者免费,同时也提供商业使用。Modeler自动生成一个基于(多个)序列比对的包含所有非氢原子的模型,用已知的相关结构建模。它以包含一个或几个结构模板序列的序列查询的对齐文件作为输入。存在不同的细化选项。经过多次周期的构建和模型评估,产生选定数量的结构模型。VHH折叠在框架中看起来很保守,但在CDR中变化很大。使用Modeller进行比较建模是从其序列中预测抗体和VHH的未知结构的合理解决方案。但是,CDR的高序列变异性使其成为一个困难的挑战。Modeller是目前在理论研究中最常用的VHH建模软件。
SWISS-MODEL
SWISS-MODEL是一个著名的在线蛋白质同源性建模服务器,首次发表于2003年的[3]。它的基础是植根于Modeller算法的原则。利用PSI-blast,结合精细的HHblits对兼容序列进行了研究。然后,用户可以选择要使用的模板。有趣的一点是,提供了不同的措施来评估所建立的几种模型的质量,即全球模型质量估计(GMQE)。结构模型的构建是通过本地开发的开放结构来完成的,即一个用于计算结构生物学的集成软件框架。以类似的方式,他们也开发了评估模型质量的措施,即QMEAN评分函数。虽然这是一种通用的方法,但它已被用于建模不同的经典抗体。SWISS-MODEL已被用于生成人源化VHH的结构模型[4],还被用于生成与SARS-COV-2棘突蛋白结合的VHH的结构模型。
Rosetta,Robetta,Rosetta Antibody and VHH Modelling Application
最初,Rosetta主要包括从头开始的方法,但逐渐地,它演变为一种从头策略,该策略结合了从数据集中的序列搜索中获得的小结构片段。一个名为Robetta的网络服务器是免费提供的(https://robetta.bakerlab.org/)[5]。
这个强大的工具也可以免费下载给学者,并可以在本地安装。针对抗体开发工具名为RosettaAntibody[6],该工具可在网上获得,并可通过Rosie (https://rosie.rosettacommons.org/antibody/)使用。该方案包括对一个给定的抗体序列的三个步骤: 1.在第一步中,使用Kabat CDR定义来识别CDR,并使用Chothia方案重新编号残基。然后对所有框架进行模板选择,以及6个CDR中的5个(CDRH1和CDRH2和CDRL1-CDRL3)。2.从选定的模板中,使用同源模型创建了一个初步的模型,因为它被证明比一个完全从头开始的方法更准确。3.CDRH3从头循环建模完成了模型预测,以及VH-VL接口的优化。
AlphaFold 2
AlphaFold是由DeepMind实验室开发的。它在2018年首次加入了CASP比赛(CASP13),并赢得了自由建模类别。两年后,通过改进,AlphaFold2极大地提高了算法的质量[7],再度赢得了CASP竞赛(CASP14)。它包含了先进的深度学习方法,再加上DeepMind的GPU计算能力,获得了巨大的成功。AlphaFold 2对应该建模的给定序列使用多序列比对(MSA)、残基配对信息和结构模板。一个名为Evoformer的变压器处理所有这些输入。关于后者的一个重要创新点是它允许在MSA和残基配对块之间交换信息。作者强调,这个Evoformer模块有助于在残基的空间和进化信息之间建立合理性。通过Evoformer后,结果信息由结构块处理,直接输出坐标。一个特殊性是中间MSA、残基配对信息和预测结构被重新注入到Evoformer中。默认情况下,这些中间结果会在网络中重新引入3次,从而改进预测。
AlphaFold2可以从GitHub存储库中下载(https://github.com/deepmind/alphafold)。但是,用户需要具有高性能GPU和大量内存的优质计算机。
另外,还可以使用在线Jupyter notebook(https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/batch/AlphaFold2_batch.ipynb)。它默认生成五个模型[8]。对于模型中的每个残差,都提供了一个置信度分数,用于对其中的模型进行排名。
RoseTTAFold和DeepAb
深度学习在结构生物信息学中发生了范式的改变。因此,著名的Rosetta利用这些新的进步已经升级。基于AlphaFold 2的特性和成功,Baker的团队广泛研究了不同的神经网络架构,并提出了一种新的基于深度学习的方法,称为RoseTTAfold [9]。它基于三轨(1D、2D和3D)神经网络,可以同时处理多个序列比对(MSA)、残基间接触和预测结构的细化。该网络中的连接允许有效地了解蛋白质序列、距离和坐标之间的关系。互连的1D和2D神经网络将裁剪的MSA和模板作为输入进行处理。3D轨道首先提供一个仅骨干模型,然后,SE(3)-transformer通过使用序列和模板之间关系的推断来执行迭代细化。到这个阶段结束时,用户将拥有一个全原子模型。采用RoseTTAfold方法的Robetta服务器为预测模型提供了一个置信度指数(每个位置的误差估计),用于对五个最终模型进行排名。
DeepAb[10]可以认为是RosettaAntibody与深度学习RoseTTAfold的自然进化。
NanoNet
NanoNet于2021年8月发表[11],这确实是第一个优化的VHH建模的方法。这种深度学习方法是使用经典抗体和VHH训练的,因为需要大量数据来训练神经网络并获得相关结果。它的架构是由一个卷积神经网络(CNN)和两个一维残差神经网络(ResNet)组成的。该程序可在线获取(https://github.com/dina-lab3D/NanoNet)。NanoNet的评估是针对AlphaFold 2进行的,2021年有16个VHH存放在PDB中,即没有参加AlphaFold 2训练。NanoNet表现更好,平均RMSD为2.69 (±1.49) ?,而AlphaFold 2为3.23 (±2.49) ?,CDR3分别为1.57 (±0.41) ?和2.04 (±2.09) ?。在使用Rosetta Antibody建模套件时,使用37个VHH获得了类似的结果。NanoNet表现更好,平均RMSD为1.68 (±0.57) ?,而Rosetta Antibody为2.71 (±1.13) ?,CDR3分别为2.99 (±1.48) ?和5.73 (±2.33) ?)。因此,NanoNet 似乎是一个非常有前途的新工具,专用于VHH建模,并取得了出色的效果。特别是,它已被用于优化预测anti-SARS-CoV-2 VHH的CDR3 [12]。
不同建模方法的测试验证
采用不同的建模方法,对已知晶体结构的VHH(PDB ID: 6XYF)进行了验证(图2)。基于6XYF的案例研究,证明NanoNet和AlphaFold 2预测了最接近实验结构的模型。当专门分析CDR3时,也发现了这种趋势。NanoNet和AlphaFold 2排名第一和第二(1.81和1.98 ?)。SwissModel(具有最佳序列同一性)在这里也表现良好,与AlphaFold 2具有相似的准确性,而其他方法的效率较低,RMSD范围在2.05和3.80 ?之间。

图2 预测VHH结构模型与PDB ID: 6XYF的X射线结构的叠加。VHH结构(绿色)与来自(a) Modeller单模板最佳模型(黄色,RMSD = 2.28 ?,RMSD CDR3 = 3.80 ?)的结构模型叠加,(b) Modeller多模板最佳模型(红色,RMSD = 2.18 ?,RMSD CDR3 = 3.32 ?),(c)ModWeb(橙色,RMSD = 1.92 ?,RMSD CDR3 = 2.05 ?),(d)具有最佳序列同一性的SwissModel最佳模型(黄色赭石色,RMSD = 1.85 ?,RMSD CDR3 = 1.98 ?),(e) 具有GMQE分数的SwissModel最佳模型(紫色,RMSD = 2.05 ?,RMSD CDR3 = 2.75 ?),(f) AlphaFold 2(小麦中色,RMSD = 1.62 ?, RMSD CDR3 = 1.98 ?), (g) RoseTTAfold (巧克力棕色, RMSD = 1.87 ?, RMSD CDR3 = 2.56 ?) 和 (h) NanoNet (蓝色, RMSD = 1.55 ?, RMSD CDR3 = 1.81 ?)。使用PyMOL进行可视化。
结 语
VHH的发现已经差不多有30年了,近年来关于VHH的研究和开发正受到越来越多的关注。利用计算机技术对VHH的建模是一种重要的研究手段,近年发展出的算法在蛋白结构预测领域获得了可喜的成果,这些算法用于VHH的建模也得到了与实验方法较为一致的结构。在本文中,根据作者初步验证,AlphaFold2和NanoNet是基于序列预测VHH结构模型的最佳工具。同时,我们也看到了目前这些算法的局限性,还有许多优化的空间。
原文链接:
[1] Vishwakarma, P.; Vattekatte, A.M.; Shinada, N.; Diharce, J.; Martins, C.; Cadet, F.; Gardebien, F.; Etchebest, C.; Nadaradjane, A.A.; de Brevern, A.G. VHH Structural Modelling Approaches: A Critical Review. Int. J. Mol. Sci. 2022, 23, 3721. https:// doi.org/10.3390/ijms23073721
[2] Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815.
[3] Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. Swiss-model: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385.
[4] Murakami, T.; Kumachi, S.; Matsunaga, Y.; Sato, M.; Wakabayashi-Nakao, K.; Masaki, H.; Yonehara, R.; Motohashi, M.; Nemoto, N.; Tsuchiya, M. Construction of a humanized artifificial vhh library reproducing structural features of camelid vhhs for therapeutics. Antibodies 2022, 11, 10.
[5] Park, H.; Kim, D.E.; Ovchinnikov, S.; Baker, D.; DiMaio, F. Automatic structure prediction of oligomeric assemblies using robetta in casp12. Proteins 2018, 86, 283–291.
[6] Sivasubramanian, A.; Sircar, A.; Chaudhury, S.; Gray, J.J. Toward high-resolution homology modeling of antibody fv regions and application to antibody-antigen docking. Proteins 2009, 74, 497–514.
[7] Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; ?ídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with alphafold. Nature 2021, 596, 583–589.
[8] Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. Colabfold-making protein folding accessible to all. bioRxiv 2022.
[9] Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876.
[10] Ruffolo, J.; Sulam, J.; Gray, J.J. Antibody structure prediction using interpretable deep learning. Patterns 2022, 3, 100406.
[11] Cohen, T.; Halfon, M.; Schneidman-Duhovny, D. Nanonet: Rapid end-to-end nanobody modeling by deep learning at sub angstrom resolution. bioRxiv 2021.
[12] Sun, D.; Sang, Z.; Kim, Y.J.; Xiang, Y.; Cohen, T.; Belford, A.K.; Huet, A.; Conway, J.F.; Sun, J.; Taylor, D.J.; et al. Potent neutralizing nanobodies resist convergent circulating variants of sars-cov-2 by targeting diverse and conserved epitopes. Nat. Commun. 2021, 12, 4676.
上一篇:国务院联防联控机制新闻发布会传递重要信息;居家时判定感染如何应对
下一篇:拥挤的120:每天呼入3.1万次,“除了猝死的和濒死的都不接送”
最近更新热点资讯
- 谷歌AI聊天记录让网友San值狂掉:研究员走火入魔认为它已具备人格,被罚带薪休假
- 豆瓣9.4,姐弟恋、三人行,这部大尺度太厉害
- Genes, Intelligence, Racial Hygiene, Gen
- 【土耳其电影】《冬眠》电影评价: 宛如一部回归伯格曼风格的道德剧
- 陌生人社会伦理问题研究
- 理论研究|前海实践的价值理性和工具理性
- 澳门刑事证据禁止规则
- 综艺普及剧本杀和密室逃脱助力线下实体店爆发式增长
- 日本小伙和五个小姐姐同居?看完我酸了!
- 第一学期高一语文考试期中试卷
- 高中必考的物理公式有哪些
- 这部大尺度的申奥片,却讲述了不lun恋...
- 心理语言学论文精品(七篇)
- 《贵妃还乡》 超清
- 专论 | 郭丹彤、陈嘉琪:古代埃及书信中的玛阿特观念
- 微专业招生 | 数字文化传播微专业列车即将发车,沿途课程抢先看!
- 生态安全的重要性汇总十篇
- 原创因“18禁”电影登舆论顶峰,万千少女一场春梦:这一生,足够了
- 章鱼头
- 读书心得体会
- 考研考北京大学医学部或者协和是一种怎样的难度?
- 央媒评女主播编造“夜宿故宫”:让肇事者付出代价,理所应当
- 库欣病患者求医记(流水账)
- 《太平公主》④ | 地位越高,越要装傻
- 爱体检 安卓版 v2.5